Protein folding models:
Models put forward to explain the phenomenon of protein folding from the random coil state described above to a native structure must predict two important features of the process for the simple case of small, single-domain proteins - two-state folding and co-operativity. These are not mutually exclusive, as a high degree of co-operativity in the folding of small proteins often leads to the observation of two-state folding. A two-state folding transition is explained simply as an equilibrium between a single folded conformation, and an unfolded state as described above, that is, the transition involves only these two states, with no accumulation of stable intermediates. The reaction co-ordinate of such a process will consist of two energy minima separated by a single energetic barrier .The folding of many small, single-domain proteins is known to follow this two-state approximation, but larger proteins tend to fold via stable intermediates.
The second important feature is that of co-operativity as determined by the experimental signature of a sigmoidal transition curve . Co-operativity between weak interactions is an important concept for much of this thesis, and is discussed further in the following chapters. Protein structures are held together purely by weak interactions - hydrogen bonds, electrostatic interactions, van der Waal's packing and hydrophobic interactions. A single weak interaction contributes very little to the stability of a protein structure, yet the very large number of these interactions act together co-operatively to impart high stability to the protein fold. Co-operativity in a folding process demonstrates that the stability of the folded state is greater than the sum of the energies of the weak interactions involved.
The first model proposed to explain the co-operativity of protein folding transitions was helix-coil theory.This represented co-operativity between neighbours in a polypeptide chain and stated that a residue has a higher propensity to be in a helical conformation if its neighbour in the sequence is also in a helical conformation. Thus, a helix can propagate quickly and co-operatively once a few adjacent amino acids adopt a helical conformation in a chance nucleation event. Although obviously limited, this theory provides a basis for much work into models for the entire protein folding process.
Subsequently, many models have been proposed to explain the protein folding problem. The ``framework model'' describes a step-wise mechanism to greatly narrow the conformational search.This involves a hierarchical assembly whereby local elements of secondary structure are formed according to the primary sequence, but independent from tertiary structure. These elements then diffuse until they collide, whereupon they coalesce to form the tertiary structure. The ``nucleation model'' suggests that tertiary structure forms as an immediate consequence of the formation of secondary structure. Nucleation occurs through the formation of native secondary structure by only a few residues (e.g. a beta-turn, or the first turn of an alpha-helix), and structure propagates out from this nucleus. The ``hydrophobic collapse model'' hypothesises that the native protein conformation forms by rearrangement of a compact collapsed structure. Hydrophobic collapse to form a molten globule therefore constitutes an early step in the folding pathway. The framework and hydrophobic collapse models suggest the formation of kinetic intermediates, whereas the nucleation model does not. Modifications of the nucleation model whereby a diffuse folding nucleus is formed and consolidated through the transition state, concomitant with tertiary structure formation leads to the ``nucleation-condensation model'' proposed by Fersht.
It is likely that folding mechanisms vary significantly according to protein size, stability and structure. The nucleation-condensation model has been supported by experimental evidence from several small proteins including chymotrysin inhibitor-II and barstar. Bychkova and Ptitsyn have studied more than 20 proteins and found that nearly all adopted a molten globule state under mild denaturing conditions. This points to the hydrophobic collapse model, a model favoured by many for the case of larger proteins.
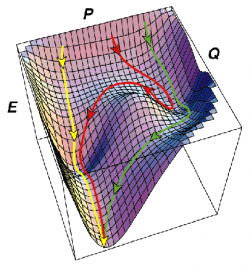
A more general description of protein folding pathways has emerged more recently termed ``folding funnel'' theory. This represents the energy surface of a protein folding pathway as a funnel, with a diverse multitude of unfolded conformations at the rim, and a single global minimum representing the native folded conformation. This leads to a ``new view'' of protein folding whereby many paths exist from the unfolded to native states, and a protein molecule may follow the steepest (fastest folding) path, or a more round-about route passing through several local minima (intermediates) and maxima (transition states).
